Das berichtet dpa am 2.8.:
"Land fördert Obduktionen von Impf- und Corona-Toten
Stuttgart (dpa/lsw) – Das Land fördert die Forschung über Langzeitfolgen einer Corona-Infektion und Obduktionen von Impf- und Corona-Toten mit 12,7 Millionen Euro. Das teilte das Wissenschaftsministerium der Deutschen Presse-Agentur mit. Es gehe um «Hilfe für Menschen, die unter Long Covid leiden, wirksamere Therapien und ein besseres Verständnis davon, warum Therapien nicht anschlagen oder Komplikationen bei Impfungen auftreten», sagte Wissenschaftsministerin Theresia Bauer (Grüne). Es brauche akut und dringend die Erkenntnisse der Wissenschaft inklusive neuer Technologien, um die Pandemie und ihre Auswirkungen zu bekämpfen.
Etwa jede vierte Patientin und jeder vierte Patient leidet nach Angaben des Ministeriums sechs bis zwölf Monate nach einer Infektion unter erheblichen Symptomen, die Gesundheit wie Arbeitsfähigkeit beeinträchtigen. Obduktionsbasierte Forschung trage zudem zum besseren Verständnis von Therapieversagen insbesondere bei neu auftretenden Varianten oder auch möglichen Impfkomplikationen bei.
An den Unikliniken im Südwesten werden bereits seit Längerem Corona-Tote obduziert, um die Erkrankung besser zu verstehen. Das Land unterstützte die Covid-Obduktionsforschung der fünf Universitätspathologien im Südwesten bereits von August 2020 bis Ende 2021 mit rund 1,8 Millionen Euro. Der Forschungsbereich werde nun erweitert, teilte das Ministerium mit. Obduktionen sollen etwa auch bei Todesfällen infolge von Impfkomplikationen durchgeführt werden. «Mehr Forschung und Transparenz tragen auch dazu bei, das Vertrauen der Menschen in die Impfung weiter zu stärken», sagte Bauer."
Zur "nebenwirkungsfreien Impfung"
ardmediathek.de (13.2.):
Siehe auch
Viele Impfnebenwirkungen nicht gemeldet? Ministerium verschleppte wichtige Datenerfassung
Kinderarzt impft„Ich werde meine jungen Patienten nicht ins offene Messer laufen lassen“
Maskenpflicht in Völlinghausen voller Erfolg: https://www.soester-anzeiger.de/lokales/moehnesee/erste-drei-waldbienenvoelker-in-station-im-wildpark-voellinghausen-91701175.html
Der Schwerpunkt dürfte doch propagandistisch auf der Obduktionen von sogenannten Corona-Toten liegen, nicht auf Impf-Toten. Es geht doch wohl eher darum, dem gemeinen Impf-Pöbel zu zeigen, wie gefährlich das Drecks-Corona ist.
Einen Lauterbach, der ideologisch-narzisstisch seit Jahren lobbyistisch aus dem Ruder läuft und stark verhaltensauffällig ist, kann kein vernünftiger Mensch ernst nehmen. Erstaunlich ist, dass sich eine ganze Gesellschaft von dieser Figur terrorisieren lässt. Die Impfung wirkt nicht, Propaganda wirkt. Die Eliten der Pharmakonzerne lachen sich schlapp.
Schlagersänger Andreas Martin sagt Tour aus "gesundheitlichen Gründen" ab: https://m.facebook.com/AndreasMartin.Musik/posts/pfbid023aAq7cESY9Qp2VtXP8M89v2R89xKr8o1patm8DkbRSc5Nd4Qz8QqeheaqudfCAfil
GeorgeOrwell3
@george_orwell3
Ob die Affenpockenimpfung tatsächlich wirkt, weiß die WHO nicht.
Jeder, dem eine Injektion verabreicht wird, wird wieder Teil einer klinischen Studie sein.…
4:39 AM · Aug 2, 2022
https://twitter.com/george_orwell3/status/1554325977274236935?cxt=HHwWjoC-ybfPiZIrAAAA
Ehrlich gesagt, vertraue ich nur Prof. Arne Burkhard mit seinem Team. Sie sind unabhängig und niemand bestimmt von außen, welche Ergebnisse opportun sind und welche nicht.
Theresia Bauer: Politik, VWL und Germanistik, Grün
Ach nee, lass mal
Jeder Vierte? Nach Alpha ider generell? Denn es hatten bekanntlich über 95% eine Omikronvariante.
Alao wieder Blödsinn.
Plötzlich und unerwartet: Nürnberger Dragqueen Uschi Unsinn bricht mit 54 Jahren tot zusammen: https://www.bild.de/lgbt/2022/lgbt/ploetzlicher-tod-mit-54-nuernberg-trauert-um-politik-dragqueen-uschi-unsinn-79157512.bild.html
Plötzlich und unerwartet: Frau stirbt bei Fallschirmsprung an "medizinischem Notfall": https://www.haz.de/der-norden/meissendorf-im-kreis-celle-fallschirmspringerin-stirbt-bei-sprung-5IP3TA4XSSMKAVKNLEIPQ474UY.html
Plötzlich und unerwartet: 27-Jaehrige Triathletin und Ärztin bricht beim Schwimmen zusammen und stirbt vier Tage später: https://uncutnews.ch/27-jaehrige-triathletin-und-aerztin-in-kanada-stirbt-vier-tage-nach-ihrem-zusammenbruch-beim-schwimmen-das-ist-der-fuenfte-todesfall-in-der-gta-innerhalb-von-2-wochen/
Zur "Obduktion". Das sagen die "Schwurbler" bereits seit über zwei Jahren (in Zahlen: 2) Sind die jetzt ungleich intelligenter, die "Schwurbler"? Und wie würden sich dann die ungleich dümmeren/dooferen bezeichnen wollen. Aber OK. Jetzt müsste es bloss noch mit Rechten Dingen zugehn. Aber, Warum nicht gleich so.
Ausserdem hätte man die gesamte Zulassung dahinter verschieben können. Der Impfstoff schützt sowieso nicht und um seine "Nebenwirkungen" wird unverschämt direkt "gefaked". Das hätte möglicherweise – absehbar – Vielen Leben und Gesundheit gerettet.
Vielen aber auch die Renditen verwehrt. Sowas
Plötzlich und unerwartet: 21-Jaehriger bricht beim Tanzen mit Gehirnblutung zusammen und stirbt: https://www.positanonews.it/2022/07/tragedia-a-palinuro-giovane-21enne-stroncato-da-un-malore-perde-la-vita-mentre-balla/3579697/
Junge Frau nach Gen-Spritze mit schrecklichen Nebenwirkungen: https://youtu.be/mw7-CrY9Rd4
@Marc Damlinger: Man kann "plötzliche und unerwartete" Fälle ja sammeln, um sie später zu überprüfen. Auf Horror-Spekulations-Videos wäre dabei gerne zu verzichten. Das ist der Stoff, mit der sachliche Kritik diskreditiert wird.
Wissenschaftliche und regulatorische Aspekte
innovativer Gentherapie-Arzneimittel
Beginn 27.09.2022 – 09:00 Uhr
Ende 27.09.2022 – 17:00 Uhr
Ort Neue Stadthalle, 63225 Langen
Veranstalter
Paul-Ehrlich-Institut
Einführung
Gentherapeutika stellen mittlerweile eine gute Behandlungsoption für Patientinnen und Patienten mit monogenen oder malignen Erkrankungen wie Lymphomen und Leukämien dar.
Innovative Ansätze wie Gen-Editierung und RNA-basierte Gentherapeutika stellen Entwickler und Regulierungsbehörden vor neue Herausforderungen.
Dieser Workshop wird einen Überblick über die regulatorischen Aspekte im Zusammenhang mit der Entwicklung innovativer Gentherapeutika geben und
versteht sich als Forum, um den offenen Austausch zwischen allen Beteiligten zu fördern.
Programmkomitee
Dr. Brigitte Anliker, Prof. Dr. Christian Buchholz, Dr. Egbert Flory, Dr. Matthias Renner, Dr. Jürgen Scherer, Dr. Martina Schüßler-Lenz
Ziele
Erleichterung des Dialogs zwischen Entwicklern und Regulatoren
Diskussion neuer Entwicklungen wie Gene Editing- und RNA-basierten Therapeutika
Darstellung neuer Herausforderungen in Bezug auf die Verordnung (EU) Nr. 536/2014 über klinische Prüfungen und die Schnittstelle zu gentechnisch veränderten Organismen (GVO)
Vermittlung von Einblicken in die wissenschaftlich regulatorischen Anforderungen im Bereich Qualität, Nicht-Klinik und Klinik für Gentherapeutika
Zielgruppe
Entwicklerinnen und Entwickler aus Akademia und Industrie (KMU, Biotech)
Medizinisches Fachpersonal
Wissenschaftliche Konsortien
Regulatorinnen und Inspektorinnen bzw. Regulatoren und Inspektoren aus nationalen Aufsichtsbehörden
Expertinnen und Experten aus der Gesundheitstechnologiebewertung und Kostenerstattung
Gebühren
Die Teilnahme an dem Workshop ist kostenlos
Weitere Informationen
Details zum Workshop-Programm und zur Anmeldung folgen in Kürze.
Der Workshop wird vom Paul-Ehrlich-Institut organisiert und als Hybridtagung durchgeführt.
https://www.pei.de/SharedDocs/veranstaltungen-events/DE/2022/2022–09-27-workshop-gentherapie-arzneimittel.html
11-jaehriges Mädchen nach zweiter Gen-Spritze mit Myocarditis auf Intensivstation: https://www.thestandard.com.hk/breaking-news/section/4/192093/Home-sweet-home-for-11-year-old-girl-battling-myocarditis
Plötzlich und unerwartet: Radrennfahrer und Nationaltrainer Jack Schiavone stirbt mit 52 Jahren: https://afipn.com.au/malta-in-mourning-as-cyclist-jack-schiavone-passes-away/
Ich weiß nicht, was diese Aufzählung bezwecken soll. Mut machen tut sie nicht, beweisen tut sie auch nichts. Eher verstärken sie diffuse Ängste, und das ist zu nichts gut.
Die Leute, die hier lesen sind sicher gut informiert und misstrauisch genug.
Was sagt die Kunst über Problematiken der Massivdeutigkeiten von Nicht-Aussagen. Die Frage ist doch ob er's nun gesagt hat, oder nicht. A "naked-mind"? – wer weiss wer weiss .…
zwar stellenweise etwas "Frauenfeindlich" auf eine besondere Art (grins), aber ein ansonsten doch "amüsantes" Werk der Filmgeschichte. Mit – wie ich seinerzeit meinte – "überraschendem" Ende. Während es – wie so oft – für Andere von Anfang an vollkommen klar war. War bestimmt dann aber auch langweiliger für Die. 🙂 (kl. Scherz)
https://youtu.be/7NhNeniXCM4?t=84
https://de.wikipedia.org/wiki/Naked_Lunch_%28Film%29
Erstes Gentherapeutikum gegen Hämophilie A erhält Zulassungsempfehlung
Der für die Bewertung von Gen-und Zelltherapien
zuständige Ausschuss für neuartige Therapien
(Committee for Advanced Therapies, CAT)
bei der Europäischen Arzneimittelagentur (European Medicines Agency, EMA) hat am 17.06.2022
für das Gentherapeutikum Roctavian
(Valoctocogene Roxaparvovec)
des US-Unternehmens BioMarin
die Empfehlung für eine
bedingte Zulassung
zur Behandlung von Erwachsenen mit schwerer Hämophilie A ausgesprochen.
Die Empfehlung des CAT wurde vom Ausschuss für Humanarzneimittel (Committee for Medicinal Products for Human Use, CHMP) bestätigt. Es handelt sich um das erste Gentherapeutikum zur Behandlung der Hämophilie A, für das in der EU eine Zulassungsempfehlung ausgesprochen wird.
Die finale Entscheidung über die Zulassung trifft die Europäische Kommission.
Erythrozyten (Quelle: qimono/pixabay.com)
Schätzungsweise 1 von 6.000 Männern von Hämophilie A betroffen
Die Hämophilie A ist die häufigste Form der sogenannten Bluterkrankheit und tritt aufgrund der zugrundeliegenden Genetik fast ausschließlich bei Männern auf.
Weil das Faktor-VIII-Gen bei Betroffenen defekt ist,
kann kein funktionsfähiges Faktor-VIII-Protein gebildet werden.
Die Häufigkeit wird im männlichen Geschlecht auf etwa 1:6.000 Personen geschätzt. Die Behandlung besteht bisher in der Substitution (dem Ersatz) des fehlenden Gerinnungsfaktors VIII, was lebenslange Injektionen erforderlich macht.
Fehlendes Faktor-VIII-Gen wird in die Leberzellen transportiert
Roctavian (Valoctocogene Roxaparvovec) ist die erste Gentherapie zur Behandlung der Hämophilie in der EU-Zulassung.
Der Wirkstoff in Roctavian ist ein AAV-Vektor (vermehrungsunfähiges Adeno-assoziiertes Virus),
das sich im Menschen nicht vermehrt und das Gen mit dem Bauplan für die Bildung des Faktor-VIII-Proteins
auf einige wenige Körperzellen überträgt.
Die Gentherapie zielt darauf ab, nach einmaliger intravenöser Gabe das Faktor-VIII-Gen in einige Leberzellen eines Patienten einzubringen, um so eine funktionsfähige Kopie des Gerinnungsfaktors zur Verfügung zu stellen.
Dies trägt dazu bei, dass Blutungen verhindert
oder Blutungsepisoden verringert werden.
Anwendung bei Patienten mit schwerer Hämophilie A
Das vom CAT empfohlene Anwendungsgebiet ist die Behandlung von Patienten mit schwerer Hämophilie A, die keine Faktor-VIII-Hemmkörper aufgrund bisheriger Faktor-VIII-Substitutionstherapie haben und
keine Antikörper gegen AAV des Serotyps 5 haben,
zu dem der in der Gentherapie verwendete AAV-Vektor gehört.
Es ist noch nicht bekannt, wie lange der Behandlungseffekt bei einem einzelnen Patienten anhalten wird.
Allerdings wurde bei der Mehrzahl der 134 in der Phase-3-Prüfung behandelten Patienten zwei Jahre nach Gentherapie eine deutliche Verringerung der Blutungen und des Verbrauchs von Gerinnungsfaktoren im Vergleich zum Vorbehandlungszeitraum beobachtet.
Bei einigen Patienten
wurde im Rahmen der klinischen Prüfung ein positiver Behandlungseffekt von bis zu fünf Jahren nach einer einzigen Infusion festgestellt.
Die Zulassungsempfehlung des CAT stützt sich auf die Ergebnisse
einer einarmigen,
nicht randomisierten
Phase-3-Prüfung an 134 männlichen Patienten mit Hämophilie A ohne Faktor-VIII-Inhibitor in der Vorgeschichte und ohne Antikörper gegen AAV 5.
Zwei Jahre nach der Verabreichung zeigten die Wirksamkeitsdaten, dass es bei der Mehrzahl der Patienten nach intravenöser Vektor-Infusion zu einem Anstieg des Faktor-VIII-Spiegels im Blut kam. Die Blutungsraten gingen um 85 % zurück und die meisten Patienten (128) benötigten keine Faktor-VIII-Ersatztherapie mehr.
Eine Erhöhung der Leberwerte ist eine aus klinischen Studien bekannte häufige Nebenwirkung von AAV-basierten Gentherapien.
Ein Anstieg des Leberenzyms Alanin-Aminotransferase (ALT) zeigt eine beginnende Leberschädigung an und wurde nach Infusion von Roctavian beobachtet.
Sie kann erfolgreich mit Kortikosteroiden behandelt werden.
Patienten mit Lebererkrankungen sollen nicht mit Roctavian behandelt werden.
Weitere häufige und vorübergehende Nebenwirkungen sind
Kopfschmerzen, Gelenkschmerzen und Übelkeit.
Um beurteilen zu können,
wie lange die Wirkung der Gentherapie anhält und
ob zusätzliche unerwünschte Wirkungen auftreten,
hat der CAT eine Nachbeobachtung der Patienten
über 15 Jahre festgelegt.
Hintergrund – Bedingte Zulassung
Wenn das klinische Datenpaket nicht als vollständig (comprehensive) betrachtet wird,
diese Daten aber nach Zulassung komplettiert werden können, ist die bedingte Zulassung (Conditional Marketing Authorisation, CMA) eine Möglichkeit,
bei einem medizinisch dringend benötigten Arzneimittel den Marktzugang zu erlauben.
Der europäische Rechtsrahmen sieht die Möglichkeit der bedingten Zulassung für Arzneimittel vor,
wenn ein ungedeckter medizinischer Bedarf (unmet medical need) besteht,
der durch das neu zugelassene Arzneimittel gedeckt wird, es um die Behandlung oder Vorbeugung von lebensbedrohlichen oder stark beeinträchtigenden Krankheiten geht und die sofortige Verfügbarkeit des Arzneimittels das Risiko aufwiegt, dass aufgrund fehlender Teilinformation zum Zeitpunkt der bedingten Zulassung bestehen könnte.
Hierbei muss sichergestellt sein, dass der Antragsteller in der Nachzulassungsphase zusätzliche Daten vorlegen kann,
die geeignet sind,
die bisher als unvollständig (non-comprehensive) erachteten klinischen Daten zu ergänzen,
um die bedingte in eine volle Zulassung zu überführen.
Hintergrund – CAT
Der CAT bei der EMA beurteilt die Qualität, Sicherheit und Wirksamkeit von Arzneimitteln für neuartige Therapien (Advanced Therapy Medicinal Products, ATMPs).
Der Ausschuss setzt sich aus Expertinnen und Experten der EU-Mitgliedstaaten auf dem Feld der ATMPs – also Gentherapeutika, Zelltherapeutika und biotechnologisch bearbeitete Gewebeprodukte – zusammen. Das Paul-Ehrlich-Institut ist Mitglied im CAT und mit Dr. Jan Müller-Berghaus, seinem Stellvertreter Dr. Egbert Flory sowie mit Dr. Martina Schüßler-Lenz als Vorsitzende des CAT vertreten.
https://www.pei.de/DE/newsroom/hp-meldungen/2022/220624-gentherapeutikum-haemophilie-a-roctavian-zulassungsempfehlung.html
aus
https://www.pei.de/DE/newsroom/hp-meldungen/hp-meldungen-node.html
Roctavian von BioMarin, d. i. Valoctocogen roxaparvovec
Patienten mit Hämophilie A können aufgrund einer Mutation im F8-Gen keine ausreichenden Mengen von normal funktionierendem Faktor VIII (Faktor 8) produzieren. Dieses essentielle Protein wird für die Blutgerinnung benötigt; ist es unzureichend vorhanden oder funktionunstüchtig, so sind die Betroffenen anfällig für stärkere und längere Blutungen, z. B. nach Verletzungen oder Operationen. Hämophilie A ist eine seltene Erkrankung, von der etwa 0,7 von 10.000 Menschen in der EU betroffen sind.
Zur Behandlung von Hämophilie A werden bislang meist Präparate eingesetzt, die den Faktor VIII enthalten, um das fehlende Protein prophylaktisch zu ersetzen. Diese Substitutionstherapie erfordert eine lebenslange ein- oder mehrmalige Gabe pro Woche oder Monat. Die Behandlung mit dem seit 2018 zugelassenen bispezifischen Antikörper Emicizumab ist als Therapiefortschritt zu sehen, erfordert jedoch auch die lebenslange regelmäßige Verabreichung alle ein bis vier Wochen. Valoctocogen roxaparvovec ist die erste Gentherapie zur Behandlung der Hämophilie A.
Valoctocogen roxaparvovec ist ein nicht replizierender, rekombinanter viraler Vektor, der auf dem Adeno-assoziierten Virus vom Serotyp 5 (AAV5) basiert und eine cDNA für die Expression der B‑Domänen-deletierten SQ-Form des humanen Gerinnungsfaktors VIII (hFVIII-SQ) unter der Kontrolle eines hybriden leberspezifischen Promoters (HLP) enthält. Der HLP regt die Transkription des Gens in sinusoidalen Endothelzellen der Leber (LSEC) an, wo auch endogener Faktor VIII hauptsächlich produziert wird.
Das ikosaedrische Viruskapsid enthält drei Strukturproteine, von denen zwei für die Kapsidbildung wesentlich sind. Das dritte ist für die Infektiosität des Kapsids bedeutsam. Es enthält eine Phospholipase A2-Domäne (PLA2-Domäne), die für den endosomalen Austritt des Kapsids und den anschließenden Transport in den Zellkern erforderlich ist.
Valoctocogen roxaparvovec wird in einem Baculovirus-Expressionssystem hergestellt, das mittels rekombinanter DNA-Technologie aus Sf-9-Zellen, einer Zelllinie der Nachtfalterart Spodoptera frugiperda, abgeleitet wurde.
Valoctocogen roxaparvovec ist ein Orphan-Arzneimittel und wurde im August 2022 unter „besonderen Bedingungen“ für die Vermarkung in der EU zugelassen (conditional approval). Das bedeutet, dass weitere Nachweise für den Nutzen des Arzneimittels erwartet werden.
de.wikipedia.org/wiki/Valoctocogen_roxaparvovec
·
Roctavian bei schwerer Hämophilie A: Anwendungsbegleitende Datenerhebung und Beschränkung der Versorgungsbefugnis beschlossen
Berlin, 2. Februar 2023 – Der Gemeinsame Bundesausschuss (G‑BA) fordert für den Einsatz des Gentherapeutikums Valoctocogen Roxaparvovec (Roctavian) eine anwendungsbegleitende Datenerhebung. (…)
Nächste Schritte
Der Hersteller von Roctavian ist nun aufgefordert, auf Basis der vom G‑BA definierten Details zur Datenerhebung das Studienprotokoll und den statistischen Analyseplan zu erstellen und dem G‑BA bis spätestens zum 2. Juli 2023 zur Abnahme vorzulegen.
Mit Start der Datenerhebung sind alle Ärztinnen und Ärzte, die das Gentherapeutikum einsetzen wollen, verpflichtet, an der Studie teilzunehmen. Ziel ist es, möglichst alle Behandlungen mit Roctavian zu erfassen und aussagekräftige Daten für die erneute Nutzenbewertung zu erhalten.
Entscheidungsgrundlagen
Grundlage des G‑BA-Beschlusses zur Forderung einer anwendungsbegleitenden Datenerhebung ist eine Recherche des Instituts für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) zu laufenden und geplanten Studien sowie das vom IQWiG erstellte Konzept. Zudem wurden Rückmeldungen aus dem Beteiligungsverfahren – zum Beispiel von den Bundesoberbehörden, den wissenschaftlich-medizinischen Fachgesellschaften, der Arzneimittelkommission der deutschen Ärzteschaft und vom betroffenen pharmazeutischen Unternehmer – berücksichtigt.
https://www.g‑ba.de/service/fachnews/55/
·
Beschluss des Bewertungsausschusses nach § 87 Abs. 1 Satz 1 SGB V in seiner 640. Sitzung am 29. März 2023
Teil A
zur Änderung des Einheitlichen Bewertungsmaßstabes (EBM)
mit Wirkung zum 1. April 2023
(…) Mit dem vorliegenden Beschluss Teil A erfolgt eine Anpassung des EBM gemäß § 87 Abs. 5b Satz 5 SGB V für den Wirkstoff Valoctocogen Roxaparvovec (Handelsname: Roctavian).
https://gkv-spitzenverband.de/media/dokumente/krankenversicherung_1/aerztliche_versorgung/verguetung_und_leistungen/beschluesse_ba_eba_de_eg/640_sitzung_ba/BA_640_Roctavian_lekto_Fassung.pdf
@ Petra S.: Geht mir genauso! Hier die Ergebnisse der Pathologie-Konferenz von Prof. Dr. Arne Burkhardt:
https://pathologie-konferenz.de/
Wer sich nach dem Anschauen noch "Impfen" lässt, dem ist sowieso nicht mehr zu helfen.
auszugsweise:
/ Nuvaxovid® – gemeldete Verdachtsfälle von Nebenwir-
kungen
in den ersten drei Monaten
seit Beginn der Imp-
fungen mit diesem Impfstoff //
Am 20.12.2021 wurde der Proteinimpfstoff Nuvaxovid® des Herstellers Novavax in der EU
für Personen ab 18 Jahren zugelassen und ist seit dem 25.02.2022 in Deutschland verfügbar.
Der Impfstoff besteht aus dem rekombinanten Spikeprotein der Ursprungsvariante des SARSCoronavirus2 (SARSCoV2) und dem saponinbasierten Adjuvans Matrix M.
Er löst im menschlichen Organismus sowohl
die Produktion neutralisierender Antikörper als
auch eine TZellantwort gegen das Spikeprotein aus.
Für eine vollständige Grundimmunisierung werden zwei Impfdosen im Abstand von drei Wochen intramuskulär verabreicht.1
Für die Zulassung wurde die Sicherheit von Nuvaxovid® anhand einer Zwischenanalyse von gepoolten Daten
aus fünf klinischen Studien
(Australien, Südafrika, Vereinigtes Königreich, USA und Mexiko) beurteilt.
Zum Zeitpunkt der Analyse hatten insgesamt 49.950 Teilnehmer
ab 18 Jahren mindestens eine Dosis Nuvaxovid® (n=30.058) oder
Placebo (n=19.892) erhalten.
Zum Zeitpunkt der Impfung lag das mediane Alter bei 48 Jahren (Bereich 18–95 Jahre).
Die mediane Dauer der Nachbeobachtung betrug 70 Tage nach der zweiten Dosis.2
In den gepoolten Reaktogenitätsdaten, die Teilnehmer ab 18 Jahren berücksichtigen, die an den zwei PhaseIIIStudien (Vereinigtes Königreich3 und USA4) teilnahmen und eine
Dosis Nuvaxovid® (n= 20.055) oder Placebo (n= 10.561) erhalten hatten, waren die häufigsten Nebenwirkungen Druckempfindlichkeit an der Injektionsstelle (75%), Schmerzen
an der Injektionsstelle (62%), Ermüdung (53%), Myalgie (51%), Kopfschmerzen (50%),
Unwohlsein (41%), Arthralgie (24%) und Übelkeit oder Erbrechen (15%).2
AKTUELLE SICHERHEITSDATEN AUS DEUTSCHLAND
696 Verdachtsfälle von Nebenwirkungen
nach Nuvaxovid®-Impfung wurden dem Paul-Ehrlich-Institut
von Verbrauchern (73%)
sowie aus medizinischen Fachkreisen (27%)
bis zum 27.05.2022
über das Spontanmeldesystem gemeldet.
Bei bis zu diesem Zeitpunkt 120.989 verabreichten Impfdosen ent
spricht dies einer
Melderate von 58 Verdachtsfallmeldungen pro 10.000 Impfdosen.
Die betroffenen Patienten waren zum Zeitpunkt des Auftretens der jeweiligen Reaktion zwischen 19 und 86 Jahre alt
(Median 43 Jahre).
Die Verdachtsmeldungen betrafen
in 507 Fällen Frauen (73%) und
in 185 Fällen Männer (27%),
wobei die Impfungen mit Nuvaxovid® sich zu 55 Prozent auf Frauen und 45 Prozent auf Männer verteilen.
In vier Verdachtsfällen fehlte die Angabe des Geschlechts.
Reaktionen traten in 58 Prozent der Fälle nach Erstimpfung und in 33 Prozent der Fälle nach Zweit-impfung auf.
In einem Fall wurden sie nach einer Boosterimpfung beobachtet, wobei hier die Grundimmunisierung mit einem anderen Impfstoff erfolgt war.
Bei neun Prozent der Meldungen wurde nicht
berichtet, um welche Impfung es sich handelt.
In 107 Fällen wurden schwerwiegende unerwünschte Reaktionen gemeldet:
42 Patienten wurden im Krankenhaus behandelt,
die übrigen Meldungen wurden als medizinisch relevant oder als unerwünschtes Ereignis von besonderem Interesse eingestuft, wenngleich formal die Kriterien für eine Klassifizie-
rung als „schwerwiegend“ nach dem Arzneimittelgesetz nicht erfüllt waren.
…
https://www.pei.de/SharedDocs/Downloads/DE/newsroom/bulletin-arzneimittelsicherheit/einzelartikel/2022-nuvaxovid.pdf
aus
https://www.pei.de/DE/newsroom/hp-meldungen/2022/220705-gemeldete-verdachtsfaelle-nebenwirkungen-nuvaxovid.html
Ohne Worte: https://corona-blog.net/2022/07/27/ploetzlich-und-unerwartet-das-unerwartete-sterben-der-50-jaehrigen-in-den-caritas-werkstaetten-am-niederrhein/
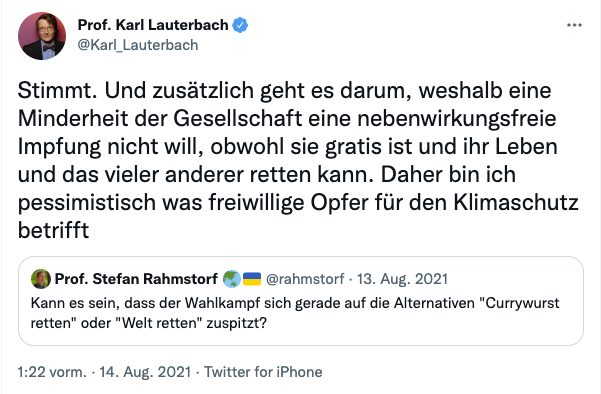
Auch wenn so ein grüner Extra-Beschluss aus BW noch keine gründliche Erfassung garantiert, zumindest ein kleines Signal (auch in Richtung von Herrn Lauterbach) dürfte es allerdings schon sein. Vielleicht dämmert es inzwischen ja auch schon mehreren Politikern, dass, wenn man schädliche Veränderungen und zunehmende Krankheiten in "Geimpften" nachweisen kann, sich solche über kurz oder lang auch in ihren eigenen Körpern breitmachen können?
Gedächtnisschwäche?
Hatte nicht die Staatsanwaltschaft im Stuttgarter Raum noch vor Kurzem Obduktionen von Menschen untersagt, die nach "Impfungen" verstorben waren?
"An den Unikliniken im Südwesten werden bereits seit Längerem Corona-Tote obduziert …"
"Der Forschungsbereich werde nun erweitert, teilte das Ministerium mit. Obduktionen sollen etwa auch bei Todesfällen infolge von Impfkomplikationen durchgeführt werden."
Jetzt will man damit beginnen, Todesfälle infolge von Impfkomplikationen zu untersuchen.
Ist also vorher nicht gemacht worden, weil nicht gefördert
Wie praktisch
«Mehr Forschung und Transparenz tragen auch dazu bei, das Vertrauen der Menschen in die Impfung weiter zu stärken», sagte Bauer."
Bei mir ist der Zug abgefahren
"Etwa jede vierte Patientin und jeder vierte Patient leidet nach Angaben des Ministeriums sechs bis zwölf Monate nach einer Infektion unter erheblichen Symptomen, die Gesundheit wie Arbeitsfähigkeit beeinträchtigen."
Was immer das bedeutet (wird hier vom Ministerium bewusst verschleiert, weil sich's gefährlicher liest, oder ist die Presse zu doof, um da nachzufragen, wer oder was genau mit "PatientInnen" gemeint ist) .
Vermutlich handelt es sich um 25% jener, die mit positivem Test entweder hospitalisiert (weswegen auch immer) oder gar
intensivmedizinisch behandelt worden waren.
Wenn es sich um die "Hospitalisierten" handelte, dann sprechen wir (laut RKI) von insgesamt knapp 600000 in den letzten 28 Monaten – also 2% von angeblich 31 Millionen "Infizierten" (Dunkelziffer nicht mitgerechnet);
– oder unterstellte 150000 – was um den Faktor 5 unter dem liegt, was uns der F***-Tor KL schon öfter erzählt
(z.B. hier:
https://www.merkur.de/deutschland/long-covid-zahlen-corona-grossbritannien-omikron-infektion-lauterbach-daten-studie-zr-91558407.html )
und wohl auf D hochgerechnet hat.
Praktischerweise lässt sich "Long Covid" hiervon nicht unterscheiden https://de.wikipedia.org/wiki/Chronisches_Ersch%C3%B6pfungssyndrom#Verbreitung
Und vom
https://de.wikipedia.org/wiki/Nocebo-Effekt
oder
https://de.wikipedia.org/wiki/Nosokomiale_Infektion
haben die Rechtgläubigen entweder noch nie etwas gehört, oder "leugnen" es einfach.
Genau. Da will dann wohl jeder auf dem Seziertisch landen. Und bitte hinten anstellen. Die Reihenfolge bestimmt der Pathologe!