Auch dieser Beitrag ist nicht auf meinem Mist gewachsen. Ein Leser (@some1) hat Folgendes gefunden: Unter der Überschrift "The European Regulatory Environment of RNA-Based Vaccines" ist 2016/2017 eine Arbeit erschienen, die Empfehlungen für die rechtliche Behandlung von mRNA-("Impf")stoffen diskutiert. In der Zusammenfassung heißt es:
»Eine Vielzahl verschiedener Arzneimittel auf mRNA-Basis befindet sich derzeit in der Entwicklung. Dies wurde möglich, da in den letzten Jahrzehnten bedeutende Durchbrüche in der RNA-Forschung beeindruckende Verbesserungen bei der Übersetzung, Stabilität und Bereitstellung von mRNA ermöglichten.
Dieser Artikel befasst sich mit Antigen-kodierenden RNA-basierten Impfstoffen, die entweder gegen Tumore oder Krankheitserreger gerichtet sind. mRNA-kodierte Impfstoffe werden sowohl zu präventiven als auch zu therapeutischen Zwecken entwickelt. Die meisten mRNA-basierten Impfstoffe werden den Patienten direkt verabreicht. Alternativ dazu werden primäre autologe Zellen von Krebspatienten ex vivo mit mRNA modifiziert und dann adoptiv auf Patienten übertragen. In der EU gibt es derzeit keine regulatorischen Leitlinien, die speziell auf mRNA-basierte Impfstoffe eingehen. Der bestehende Rechtsrahmen legt jedoch eindeutig fest, dass mRNA-basierte Impfstoffe in den meisten Fällen zentral zugelassen werden müssen. Interessanterweise werden RNA-basierte Impfstoffe, je nachdem, ob sie gegen Tumore oder Infektionskrankheiten gerichtet sind, formell als Gentherapieprodukte betrachtet oder nicht. Neben einem Überblick über den aktuellen klinischen Einsatz von mRNA-Vakzinen in verschiedenen Therapiegebieten wird die aktuelle regulatorische Situation detailliert erörtert und es werden regulatorische Perspektiven diskutiert.«
Den eigentlichen Aufsatz gibt es hier. Zu den VerfasserInnen gehören die MarktteilnehmerInnen Ugur Sahin und Özlem Türeci.
Wer ist Dr. Thomas Hinz?
Der Korrespondenzautor ist Thomas Hinz vom Paul-Ehrlich-Institut, der bereits 2006 gemeinsam mit dem PEI-Chef Klaus Cichutek zu diesem Thema veröffentlichte (Manufacturing and quality control of cell-based tumor vaccines: a scientific and a regulatory perspective).
Aus dem Jahr 2016 stammt eine Präsentation von Hinz unter dem Logo des Paul-Ehrlich-Instituts für die EMA. Dort stellt er die Frage
»Zu welcher Klasse von Medikamenten gehört die mRNA?
Rechtliche Definition von Gentherapie (RICHTLINIE 2009/120/EG):
Unter einem Gentherapeutikum ist ein biologisches Arzneimittel zu verstehen, das folgende Merkmale aufweist:
-
-
-
- Es enthält einen Wirkstoff, der eine rekombinante Nukleinsäure enthält oder daraus besteht, der im Menschen verwendet oder ihm verabreicht wird, um eine Nukleinsäuresequenz zu regulieren, zu reparieren, zu ersetzen, hinzuzufügen oder zu entfernen.
- Seine therapeutische, prophylaktische oder diagnostische Wirkung steht in unmittelbarem Zusammenhang mit der rekombinanten Nukleinsäuresequenz, die es enthält, oder mit dem Produkt, das aus der Expression dieser Sequenz resultiert.
-
-
Impfstoffe gegen Infektionskrankheiten sind keine Gentherapeutika.«
(Der deutsche Text der Richtlinie wird hier zitiert nach eur-lex.europa.eu.)
Hinz kann sich also auf eine willkürliche Festlegung der EU-Kommission stützen. Er zeigt zunächst ein Schaubild von Ugur Sahin unter einer Überschrift, die das Gegenteil nahelegt:
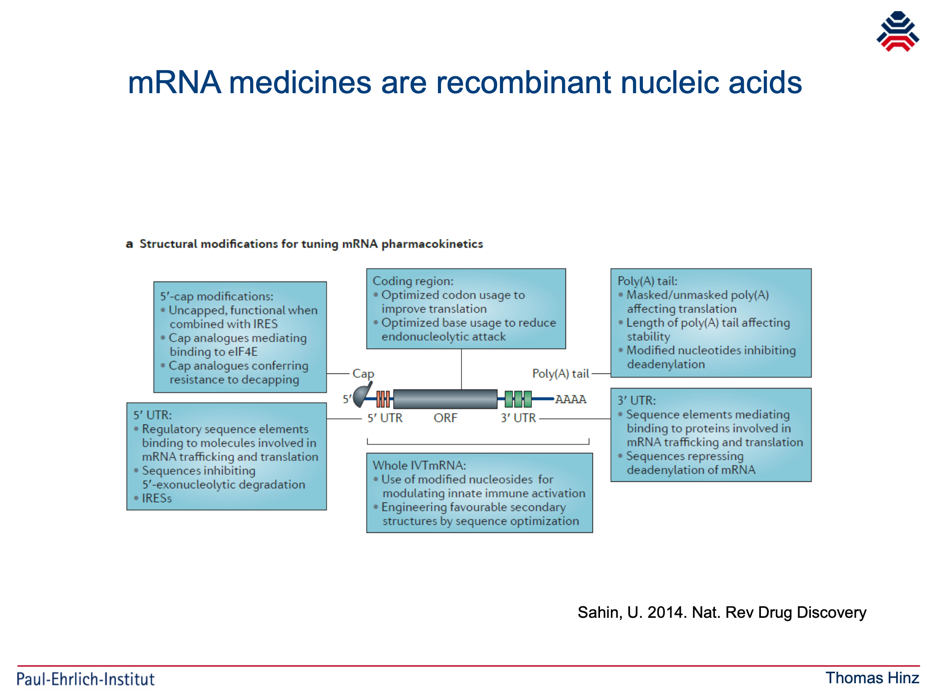
Hinz fährt fort:
»Ist rekombinante mRNA ein biologisches Arzneimittel?
-
-
-
- Unter anderem
- Arzneimittel, die mit einem der folgenden biotechnologischen Verfahren entwickelt wurden:
- rekombinante DNA-Technologie...
- Arzneimittel, die mit einem der folgenden biotechnologischen Verfahren entwickelt wurden:
- Unter anderem
-
-
-
-
-
- Schlussfolgerung
- Chemische DNA-Rekombination/Synthese einer Vorlage, gefolgt von einer in vitro mRNA-Transkription, ergibt wahrscheinlich kein biologisches Arzneimittel → keine Gentherapie
- Die Verwendung rekombinanter Plasmide als biotechnologisch erzeugte Vorlagen, an denen Bakterien beteiligt sind, führt wahrscheinlich zu einem biologischen Produkt → Gentherapie «
- Schlussfolgerung
-
-
Was der Mitarbeiter des Paul-Ehrlich-Institus im Rahmen dieser Präsentation nur skizziert, ist exakt das, was er in oben genanntem Aufsatz gemeinsam mit Sahin und Türeci entwickelt. Dort heißt es ausführlicher:
»Die Produktion von Proteinen unter GMP-Bedingungen ist mit einem erheblichen Ressourcen-, Kosten- und Zeitaufwand verbunden. Die Verwendung von DNA oder mRNA für die Expression praktisch aller Proteine in vivo erscheint daher als attraktive Alternative zur Ex-vivo-Produktion von Proteinen, wodurch der Organismus im Wesentlichen zu seiner eigenen Produktionseinheit wird. mRNA erscheint daher als günstiger Vektor für die Produktion weithin benötigter Proteine wie Antigene oder Proteine...«
GMP steht für Gute Herstellungspraxis (Good Manufacturing Practice). Zum Verfahren wird ausgeführt:
»Entwickelte mRNA-Moleküle können auch für die In-vitro-Transfektion von Zellen verwendet werden. Da die spontane mRNA-Aufnahme je nach Zelltyp sehr gering ist, sind verschiedene Transfektionsmethoden erforderlich. Interessanterweise führt auch die direkte Injektion von mRNA in die Zelle zu einer erheblichen Proteinexpression. In letzter Zeit wurde auch die intravenöse Verabreichung von in Liposomen verpackter mRNA erfolgreich eingesetzt. Die liposomalen Nanopartikel schützen die mRNA vor dem sofortigen Abbau durch Serum-RNasen und sind außerdem so konstruiert, dass sie die mRNA gezielt an bestimmte Organe weiterleiten...
Derzeitige Krebsimpfstoffe werden wiederholt und in Situationen verabreicht, in denen vermutlich eine hohe Antigenlast vorhanden ist.
Impfstoffe gegen Infektionskrankheiten hingegen werden gesunden Menschen prophylaktisch verabreicht, um mit möglichst wenigen Verabreichungen eine schützende Immunreaktion auszulösen. Dies stellt neue Anforderungen an die Sicherheit und die Induktion einer schützenden Immunreaktion... Ein weiteres wichtiges Merkmal der mRNA-basierten Vakzinologie ist, dass diese Technologie die schnelle Entwicklung neuer Impfstoffe innerhalb einer sehr kurzen Zeitspanne von Wochen statt Monaten ermöglicht... Die Entwicklung von Impfstoffen könnte also durch mRNA-basierte Impfstoffe revolutioniert werden...«
Chemisch hergestellte Gentherapeutika sind keine mehr
Langsam kommen die AutorInnen zum Kern ihrer Überlegungen. Zunächst wird das übliche Verfahren der Zulassung beschrieben (s. ausführlich unten). Ausgehend von zitierter Richtlinie heißt es:
»Aus dieser Definition lassen sich die folgenden wichtigen Schlussfolgerungen ziehen: Da der übliche Herstellungsprozess von mRNAs auf der In-vitro-Transkription mit aus Bakterien gewonnenen Plasmidvorlagen beruht, sind solche mRNAs vermutlich als biologisches Arzneimittel zu betrachten. Außerdem sind mRNAs in der Regel rekombinant, da verschiedene Modifikationen wie Codon-Optimierung, modifizierte CAP-Strukturen, Einführung geeigneter 5′- und 3′-nichtkodierender Regionen, definierte Poly(A)-Schwänze usw. vorgenommen werden. Zusammengenommen werden mRNAs, die die Kriterien eines rekombinanten biologischen Produkts erfüllen, das zum Hinzufügen oder Ersetzen einer genetischen Sequenz verwendet wird und dessen therapeutische, prophylaktische oder diagnostische Wirkung direkt durch die darin enthaltene Nukleinsäure vermittelt wird, als Gentherapeutika definiert. Sollte ein RNA-Molekül auf rein chemischem Wege hergestellt werden, wie es bei vielen RNAi-Molekülen der Fall ist, wäre es kein biologisches Produkt mehr und könnte daher nicht als Gentherapeutikum eingestuft werden.
Wichtig ist auch der Hinweis, dass eine mRNA zur Behandlung oder Vorbeugung von Infektionskrankheiten per Gesetz kein Gentherapieprodukt ist, auch wenn alle anderen Anforderungen erfüllt sind (rekombinant, biologisch). Folglich ist ein mRNA-Molekül, das zur prophylaktischen Impfung, z. B. gegen Grippe, verwendet wird, kein Gentherapieprodukt, während dies beispielsweise bei der Behandlung von Krebs der Fall ist. Für die EMA-Zulassung hat dies zur Folge, dass mRNA zur Impfung gegen Infektionskrankheiten vom CHMP bewertet wird, während mRNAs, die die Kriterien eines ATMP erfüllen, vom CAT bewertet werden.«
ATMP = Advanced Therapy Medicinal Products
Alles eine Frage der Definition
Mit welchen Tricks die Klassifizierung eines Produkts als gentherapeutisch und damit eine strengere Prüfung durch CAT (Committee for Advanced Therapies der EMA) verhindert wird, zeigt dieses Beispiel:
»Die Bedeutung von "... seine therapeutische, prophylaktische oder diagnostische Wirkung bezieht sich unmittelbar auf die rekombinante Nukleinsäuresequenz, die es enthält, oder auf das Produkt der genetischen Expression dieser Sequenz", wie unter Buchstabe a) der Legaldefinition dargelegt, wird durch die CAT-Klassifizierungen von T-Zellen beleuchtet, die genetisch so verändert wurden, dass sie ein exogenes Thymidinkinase (TK)-Gen exprimieren. Die T-Zellen wurden nicht als Gentherapeutikum eingestuft, da sie für die Immunrekonstitution nach einer hämatopoetischen Stammzelltransplantation bestimmt waren. Der Zweck des eingeführten TK-Gens bestand darin, das Auftreten einer Transplantat-gegen-Wirt-Krankheit zu behandeln, sollte diese bei bestimmten Patienten auftreten. Die in den Patienten eingebrachte Gensequenz (das TK-Gen) stand also nicht in direktem Zusammenhang mit der beabsichtigten therapeutischen Wirkung, d. h. der Immunrekonstitution. Die Zellen wurden daher als somatisches Zelltherapieprodukt eingestuft. Bei T-Zellen hingegen, die mit mRNA transfiziert sind, die z. B. für einen neuen T-Zell-Rezeptor (TCR) kodiert, steht der neu eingeführte TCR eindeutig in direktem Zusammenhang mit der beabsichtigten therapeutischen Wirkung, d. h. der Abtötung von Krebszellen, die das von einem solchen TCR erkannte Zielantigen exprimieren. In diesem Szenario würde das Arzneimittel wahrscheinlich als ATMP eingestuft werden. Solche Ansätze können für die Entwicklung adoptiver zellulärer Therapien gegen neuartige Targets attraktiv sein, für die keine ausreichenden Sicherheitsdaten vorliegen, die aber aufgrund des vorübergehenden Charakters der genetischen Veränderung ein akzeptables Risiko-Nutzen-Profil aufweisen.«
Das Problem: Vorbeugung von durch die Infektion ausgelösten Krankheiten
»Wie oben dargelegt, gehören Impfstoffe gegen Infektionskrankheiten nicht zu den Gentherapie-Arzneimitteln. Im CAT-Reflexionspapier wird jedoch darauf hingewiesen, dass ein gentherapeutischer Impfstoff dennoch als Gentherapie eingestuft werden kann, wenn er zur Behandlung oder Vorbeugung von durch die Infektion ausgelösten Krankheiten (z. B. Malignome) indiziert ist. So ist beispielsweise ein mRNA-basierter Impfstoff zur Behandlung oder Vorbeugung von HPV16-induzierten malignen Erkrankungen ein Gentherapieprodukt (wenn die Kriterien für Gentherapie erfüllt sind). Wird die identische mRNA für die Impfung gegen HPV16 verwendet, so wird sie als Impfstoff eingestuft...
Die EMA hat bisher noch keine spezifischen Leitlinien für die Entwicklung von mRNA-basierten Arzneimitteln veröffentlicht. Daher müssen die allgemeinen Grundsätze befolgt werden, die in übergreifenden Leitfäden dargelegt sind. Obwohl es sich bei mRNA-basierten Impfstoffen zur Vorbeugung oder Behandlung von Infektionskrankheiten nicht um gentherapeutische Produkte handelt, sollten die in der EMA-Leitlinie für gentherapeutische Arzneimittel dargelegten Grundsätze beachtet werden, die sich mit Qualität, nichtklinischen und klinischen Aspekten befassen...«
Die Achillesferse der heutigen "Impfstoffe"
Hier könnte eine erhebliche Schwierigkeit für die heute verwendeten "Impfstoffe" liegen. Da sie der herkömmlichen Definition von Impfstoffen nicht entsprechen – sie erzeugen weder eine Immunität noch verhindern sie Übertragungen –, könnte der oben beschriebene Fall angenommen werden. Wenn das Hauptmerkmal die Vermeidung schwerer Verläufe ist, wird man von einer "Vorbeugung von durch die Infektion ausgelösten Krankheiten" sprechen können. Damit läge wiederum eine Gentherapie vor, für die es aber keine Prüfung und damit Zulassung gab.
Gar nicht gut für Sahin und Türeci klingt auch der nächste Absatz.
mRNA könnte im Plasma verbleiben und proinflammatorische Zytokinen induzieren
»Es gibt keine spezielle Leitlinie, die sich mit der präklinischen Bewertung von mRNA-Impfstoffen befasst. Die präklinische pharmakologische und toxikologische Bewertung von mRNA hängt weitgehend von der zu behandelnden Krankheit und der Art der Verabreichung ab. Das folgende Beispiel mag diese Tatsache verdeutlichen. Impfstoffe werden in der Regel lokal über den intradermalen oder intramuskulären Weg verabreicht... Bei einmaliger oder wiederholter systemischer Verabreichung scheint die Bewertung der pharmakokinetischen Parameter sowohl unter Sicherheits- als auch unter Wirksamkeitsaspekten wichtig zu sein. Relevante Plasma-PK-Parameter bei systemischer Verabreichung sind Exposition, Clearance und Akkumulation. Aufgrund der erhöhten Stabilität, die durch die Komplexbildung mit geeigneten Substanzen erreicht wird, könnte sich die mRNA bei wiederholter Verabreichung über einen gewissen Zeitraum im Plasma anreichern oder dort verbleiben. Dies wiederum könnte bedenklich sein, da mRNA per se immunstimulierend wirkt und die Sekretion von proinflammatorischen Zytokinen induzieren kann.«
Aktuell wirbt Hinz für die dieses Event:
Das vorläufige Programm gibt es hier.
Wer ist Kajo Kallen?
Ein weiterer Autor des Artikels – neben Cedrik M Britten und Axel Hoos vom Pharmakonzern Glaxo Smith Kline – ist Kajo Kallen, der hier für die Kallen Consulting in Frechen aufgeführt ist. Es könnte sich dabei um Karl-Josef Kallen handeln, ehemals Chief Marketing Officer der Firma eTheRNA. Sie stellt sich so vor:
»Freisetzung des gesamten Potenzials der mRNA-Technologie
... eTheRNA wurde im Januar 2013 als Spin-off-Unternehmen der Vrije Universiteit Brussel (VUB) nach der Entwicklung seiner TriMix mRNA-Technologie gegründet.«
Eine Kajo Kallen präsentierte im Oktober 2010 für die Firma Curevac, die damals auf der Paul-Ehrlich-Str. 15 in Tübingen residierte, diesen Erfolg:
»CureVac präsentiert überzeugende Daten zu der weltweit ersten klinischen Phase-I/IIa-Studie mit einer mRNA-Vakzine
... Tübingen, 04. Oktober 2010. Die CureVac GmbH, ein auf mRNA-Vakzine spezialisiertes Unternehmen, gab heute die ersten Daten einer nicht-verblindeten klinischen Phase-I/IIa-Studie mit ihrem mRNA-Vakzin CV9103 bei Patienten mit hormonunempfindlichem (hormonrefraktärem) Prostatakrebs bekannt...
Diese Daten sind äußerst ermutigend", sagte Dr. Kajo Kallen, CSO von CureVac. "Die Ergebnisse bedeuten eine erste wichtige Validierung von CureVacs innovativer firmeneigener RNActive(®)-Impfstoff-Technologie am Menschen."...
Dr. Ingmar Hoerr, Geschäftsführer von CureVac, fügte hinzu: "Diese ersten Ergebnisse bedeuten exzellente Neuigkeiten für CureVac. Wir sind hoch motiviert, die Leistungsfähigkeit unserer RNActive(®)-Impfstoff-Technologie in den Indikationen Krebs und Infektionskrankheiten weiter unter Beweis zu stellen."...«
firmenpresse.de (4.10.2010)
Nachtrag aus dem Papier zum normalen Zulassungsverfahren:
»EMA und nationale Aufsichtsbehörden
Wie jedes andere Arzneimittel werden auch mRNA-basierte Arzneimittel in der EU je nach ihrem Entwicklungsstadium sowohl auf nationaler als auch auf EU-Ebene reguliert. Während die Durchführung klinischer Studien einschließlich der Herstellung von Prüfpräparaten auf nationaler Ebene geregelt ist, werden mehrere Produkte auf die EU-Ebene verlagert, sobald eine Marktzulassung beantragt wird.
Je nach Produktklasse kann ein mRNA-Arzneimittel eine Zulassung in allen EU-Mitgliedstaaten erhalten, indem es das so genannte zentralisierte Verfahren durchläuft. Die Anträge auf Marktzulassung werden vom Ausschuss für Humanarzneimittel (CHMP) bei der Europäischen Arzneimittelagentur (EMA) geprüft. Das Gutachten des CHMP über die Genehmigungsfähigkeit eines Arzneimittels wird an die Europäische Kommission weitergeleitet. Die Europäische Kommission ist somit die letzte Institution, die einen Zulassungsantrag bewilligt oder ablehnt.
Antragsteller wie Biotechnologie- oder Pharmaunternehmen müssen der EMA ein Dossier übermitteln, das die Herstellung und Qualitätskontrolle sowie die präklinischen und klinischen Studien umfassend beschreibt. Sobald das zentralisierte EMA-Verfahren eingeleitet ist, folgt es einem festgelegten Zeitplan, der eine eingehende Bewertung der eingereichten Daten vorsieht, die in eine Liste von Fragen mündet, die dem Antragsteller spätestens 120 Tage nach dem Tag der Einleitung des Verfahrens zugesandt wird. Die Bewertung erfolgt getrennt durch einen Berichterstatter und einen Mitberichterstatter, die vom CHMP benannt werden. Im CHMP sind Vertreter aller Zulassungsbehörden der EU-Mitgliedstaaten vertreten und bilden somit ein großes Netzwerk europäischer Zulassungsbehörden. Der Fragenkatalog für den Tag 120 wird vom CHMP nach Erörterung der getrennten Beurteilungen des Berichterstatters und des Mitberichterstatters, nach Erhalt von Kommentaren und Beiträgen der CHMP-Mitglieder/nationalen Agenturen und nach Bewertung des Pharmakovigilanz- und Risikomanagementsystems durch den Ausschuss für Risikobewertung im Bereich der Pharmakovigilanz (PRAC) der EMA angenommen.
Nach der Übermittlung des Fragenkatalogs an den Antragsteller an Tag 120 wird die Frist (aus Sicht der EMA) um etwa 90 Tage verlängert. Während dieser Zeit hat der Antragsteller die Möglichkeit, eine Antwort zu verfassen. Nach Eingang der Antworten des Antragstellers auf jede gestellte Frage wird das Verfahren bei der EMA am Tag 121 wieder aufgenommen. Die Bewertung der Antworten durch den Berichterstatter, den Mitberichterstatter, die CHMP-Mitglieder und den PRAC wird am Tag 180 mit der Verabschiedung einer Liste der noch offenen Fragen abgeschlossen, die dem Antragsteller erneut übermittelt wird. Es folgt eine weitere Unterbrechung (1-3 Monate), die es dem Antragsteller ermöglicht, Antworten an die EMA zu übermitteln, was zur Wiederaufnahme des Verfahrens am Tag 181 führt. Das endgültige Gutachten über die Erteilung einer Genehmigung für das Inverkehrbringen wird vom CHMP am Tag 210 abgegeben. Dieses CHMP-Gutachten wird an die Europäische Kommission weitergeleitet, die am Tag 277 endgültig entscheidet. Die Antragsteller sollten sich darüber im Klaren sein, dass die Zulassungsanträge auch die Ergebnisse der im pädiatrischen Prüfkonzept (PIP) beschriebenen Studien enthalten müssen, es sei denn, das Arzneimittel wurde von dieser Anforderung freigestellt. Der Pädiatrieausschuss (PDCO) der EMA ist für die Genehmigung oder Ablehnung des PIP zuständig.
Neben dem 210 Tage dauernden EMA-Verfahren können die Antragsteller ein beschleunigtes Verfahren beantragen, das nur 150 Tage dauert. Das beschleunigte Verfahren ist anwendbar, wenn das Arzneimittel sowohl aus Sicht der öffentlichen Gesundheit als auch aus Sicht der therapeutischen Innovation von großem Interesse ist.
Neben dem oben beschriebenen "normalen" Weg der zentralisierten Zulassung durch die EMA auf der Grundlage umfassender Daten ist es auch möglich, Produkte auf der Grundlage unvollständiger Daten zuzulassen. Eine solche bedingte Zulassung kann erteilt werden, wenn nur vorläufige klinische Sicherheits- und Wirksamkeitsdaten vorliegen. Auch wenn die klinischen Daten unvollständig sind, muss das von der EMA im Rahmen des Bewertungsverfahrens ermittelte Nutzen-Risiko-Verhältnis insgesamt positiv ausfallen. Eine weitere Voraussetzung für diesen alternativen Zulassungsweg ist die Fähigkeit des Antragstellers, die fehlenden Daten nach Erteilung der bedingten Zulassung nachzureichen. Außerdem muss das Arzneimittel für Patienten mit ungedecktem medizinischem Bedarf bestimmt sein, und der Nutzen für die öffentliche Gesundheit muss die Risiken überwiegen, die mit den unvollständigen klinischen Daten verbunden sein könnten. Nach Vervollständigung der Daten kann die bedingte Zulassung in eine reguläre Zulassung umgewandelt werden...
Verordnungen sind ab ihrem Inkrafttreten für alle EU-Mitgliedstaaten rechtsverbindlich und müssen nicht wie Richtlinien in die Gesetzgebung der Mitgliedstaaten umgesetzt werden...«
(Hervorhebungen nicht in den Originalen.)
Der Fund ist gut. Der Fallout dieser Plandemie wird – hoffentlich – die ganze Pharma-Industrie und die Aufsichtsbehörden aufrollen.
Gesamtmetall bekommt kalte Füße
https://www.fr.de/wirtschaft/metall-elektro-industrie-deutschland-corona-impfpflicht-betretungsverbot-warnung-gesamtmetall-bundestag-zr-91261944.html
"Wolf betonte bei „Bild TV“ am Dienstag weiter, es werde für die Branche „ganz schwierig, wenn das Betretungsverbot kommt – wie wir es in der Pflege schon haben.“ Viele Unternehmen könnten dann nicht mehr produzieren, Lieferketten würden unterbrochen: „Die Automobilindustrie ist sehr arbeitsteilig. Viele Zulieferer gehören dazu. Das käme alles zum Erliegen.“
Die Chip-Krise sei schon schlimm genug. „Wenn jetzt durch einen Impfzwang noch zusätzliche Unterbrechungen kämen, würde Deutschland kein einziges Auto mehr produzieren.“ Es gebe 700.000 bis 800.000 Mitarbeiter, die nicht geimpft sind, schätzte Wolf. Insgesamt gibt es in der Metall- und Elektroindustrie in Deutschland* 3,9 Millionen Beschäftigte. "
falls ich richtig Dr. Wodarg verstanden haben, der Bundestag soll schon damals die jetzige Operation gesetzlich vorbereiten. Vodarg, ehem. SPD-Politiker soll damals, 2009, nicht die drohende Gefahr erkannt haben.
Die Pläne uns mit genetischer „Impfung“ (und was noch dazu ?) zu verseuchen sind älter als 10 Jahre, vielleicht sogar 20, in diesen Dimensionen wird unser Schicksal von den Milliardären geplant, der Bundestag ist nur ein ihrer Gehilfe.
- https://www.handelsblatt.com/politik/konjunktur/nachrichten/entwicklungshilfeorganisation-oxfam-bericht-zehn-reichste-menschen-konnten-vermoegen-in-der-pandemie-verdoppeln/27980442.html
oder in gleichem Medium:
„Vermögensverteilung Superreiche steigern ihr Vermögen in der Krise um mehr als 50 Prozent “ in einem Jahr.
(akac)
@ Seit 2009 …
Ich empfehle Ihnen die Recherche der TNI zur Infiltration der UN und ihrer Mitgliedsstaaten:
https://www.tni.org/files/publication-downloads/great_takeoverbook_-_14_jan_2022.pdf
Die TNI ist selbst eine NGO in diesem Umfeld, allerdings eine der eher linken, denen die Bereicherung und Einflussnahme, die sie selbst vor Ort im Lauf der Jahre beobachten, ein Dorn im Auge ist:
https://www.tni.org/en/article/davos-and-its-danger-to-democracy (2016)
https://www.tni.org/en/publication/the-great-takeover
Am 13.12.2021 hat sich zudem die WHO zu folgendem entschlossen:
WHO reform
Involvement of non-State actors in WHO’s governing bodies
https://apps.who.int/gb/ebwha/pdf_files/EB150/B150_37-en.pdf
Es wird immer deutlicher, dass JETZT zunehmend auch kein Geheimnis mehr daraus gemacht werden soll, sondern die Übernahme offen erfolgt.
Warum und wie ist das möglich?
Es ist eigentlich ziemlich einfach: die Politiker der Welt sind ratlos, wie sie die Probleme lösen soll, die die Agenda 21 ihnen aufgibt. Die Philanthropen hingegen haben Pläne, Vorstellungen und Techniken, wie das geht und sie sagen den Politikern, dass sie ihnen dabei helfen werden, dass sie die Mittel haben. Dankbar strahlend nehmen Politiker das Angebot an, selbst keine Lösungen mehr finden zu müssen, sondern selbst nur noch umzusetzen und dafür auch noch belohnt zu werden – längst nennt man es nicht mehr Bestechung, sondern sie werden ja für "das Gute" belohnt, das sie tun dürfen.
Was damals wie heute fehlt, sind die eigenen Pläne und die Einigkeit darüber. Wenn die WEF-Leute "weg" wären, wüsste keiner, was zu tun ist. Das trifft auch auf den Widerstand zu, der es in zwei Jahren nicht fertig gebracht hat, eine konzise Analyse UND ebensolche eigenen Vorstellungen zu entwickeln, die man dem Great Takeover entgegen halten kann. Der einzige Feind einer schlechten Lösung ist jedoch eine bessere!
Wodarg ist kein "Übermensch", er hat damals nicht alles bemerkt, ja. Sie und ich und all die anderen aber auch nicht! Die Vorstellung dessen, was sich gerade vollzieht, war kaum einem möglich. Es gab immer nur zwei "Sorten" von Autoren, die sich damit beschäftigten: Verschwörungstheoretiker und die Akademiker der Think Tanks. Beides konnte offen geschehen und ist gratis abrufbar, denn: den einen hat man nicht geglaubt, die anderen hat man nicht gelesen.
So ist es aber bis heute! Wer begreift, was die EU seit letztem Sommer gesetzlich für alle Mitglieder ungehindert in Kraft setzt, dem muss übel werden! Aber: es tut keiner. KEINER.
Nee, die Pharma-Kampagne um chemische Präparate und "Impfungen" als über jede Natürlichkeit erhaben und wirkungsvoller darzustellen begann schon in den Neunzehnhundertzwanzigern mit Herr Rockefeller. Er wollte sein schönes Erdöl noch besser und teurer Vermarkten als nur als Treibstoff. STP, "Scientifically Treated Petroleum", versprach wesentlich bessere Gewinnmargen als Benzin.
"The Rockefeller Foundation is an American private foundation based at 420 Fifth Avenue, New York City.[3] It was established by the Rockefeller family in New York State on May 14, 1913, "
@aa
Auch Ihr Beitrag zum Thema ist herausragend gut!
Danke für die Recherche.
Tim Roehn und Norbert Härung werden sich freuen für die Vorarbeiten von Ihnen und some1!
Es ist sehr durchsichtig. Die EMA hat mit Datum vom 19.2.21 ihren "Public Assessment Report" zu Comirnaty korrigiert, um klarzustellen, dass sie auf Umweltverträglichkeitsprüfung/Environmental Risk Assessment (bei Gentherapeutika ohne Ausnahme vorgeschrieben) wegen einer CHMP-Leitlinie von 2006 verzichtet hat, der zufolge Impfstoffe generell kein Risiko für die Umwelt darstellen, Im Wortlaut (übersetzt mithilfe von deepl): "Gemäß der CHMP-Leitlinie zur Umweltverträglichkeitsprüfung von Humanarzneimitteln (EMEA/CHMP/SWP/4447100 Corr 2) ist es aufgrund ihrer Beschaffenheit unwahrscheinlich, dass Impfstoffe und Lipide zu einem signifikanten Risiko für die Umwelt führen. Daher werden in diesem Zulassungsantrag keine Studien zur Umweltrisikobewertung vorgelegt, was als akzeptabel angesehen wird."
Original: "In accordance with the CHMP Guideline on the Environmental Risk Assessment of Medicinal Products for Human Use (EMEA/CHMP/SWP/4447100 Corr 2), due to their nature vaccines and lipids are unlikely to result in a significant risk to the environment. Therefore, environmental risk assessment studies are not provided in this Application for Marketing Authorisation, which is considered acceptable."
https://www.ema.europa.eu/en/documents/assessment-report/comirnaty-epar-public-assessment-report_en.pdf
Die Schweizer Zulassungsbehörde, welche sich auf die EMA-Zulassung stützt, erwähnt in ihrem PAR Comirnaty ERA nur einmal – im Abkürzungsverzeichnis . Sie geht mit keinem Wort darauf ein, warum ERA nicht durchgeführt wurde.
Zeit für eine Forensik-Serie?
1.
"GMP steht für Gute Herstellungspraxis (Good Manufacturing Practice)."
Die nach Feststellung der og. Autoren unzureichenden Vorlagen für GMP, die EMA zur "Prüfung" verwendet, werden von einer PPP namens "The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) " geliefert.
Die EMA berichtet:
Die ICH erreicht die Harmonisierung durch die Entwicklung von Leitlinien und technischen Anforderungen für die Entwicklung, Zulassung und Sicherheitsüberwachung von Arzneimitteln unter Einbeziehung von Experten aus Behörden und Industrie. Die Mitglieder der ICH nehmen die Leitlinien an und sind verpflichtet, sie umzusetzen. (übersetzt deepl.com)
https://www.ema.europa.eu/en/partners-networks/international-activities/multilateral-organisations-initiatives/international-council-harmonisation-technical-requirements-registration-pharmaceuticals-human-use
Hier die Mitgliederliste:
https://www.ich.org/page/members-observers
Neben der EMA sind die FDA und die japanische Aufsichtsbehörde dort Gründungs-Mitglied.
Man findet dort neben der BMGF und CIOMS so ziemlich alle denkbaren Regulierungsbehörden, als weitere Mitglieder die ALLE die (gleichen) Vorlagen der ICH verwenden! Das gilt auch für die WHO, die gemeinsam mit dem Industriepartner als Observer fungiert.
Auf der Industrieseite der ICH stehen Mitglieder der International Federation of Pharmaceutical Manufacturers and Association (IFPMA), die die ICH gegründet hat:
Seit ihrer Gründung im Jahr 1990 hat die International Federation of Pharmaceutical Manufacturers and Associations (IFPMA) das Sekretariat der ICH-Organisation gestellt und war als nicht stimmberechtigtes Mitglied im ICH-Lenkungsausschuss vertreten, der alle Harmonisierungsaktivitäten überwachte. IFPMA war auch als Treuhänder der ICH tätig.
Am 23. Oktober 2015 wurde die ICH als internationaler gemeinnütziger Verein nach Schweizer Recht gegründet, für den die IFPMA gemäß Artikel 16 der ICH-Statuten als ständiger Beobachter und gemäß Artikel 27 Absatz 4 als ständiger Beobachter des Verwaltungsausschusses benannt wurde.
Übersetzt mit http://www.DeepL.com/Translator (kostenlose Version)
https://www.ifpma.org/wp-content/uploads/2017/06/ICH-Terms-of-Reference_REVISED_18Jul2018_updated.pdf
"Governance and Leadership der IFPMA" sehen derzeit so aus:
• Jean-Christophe Tellier, IFPMA President and Chair of the IFPMA CEO Steering Committee, Chief Executive Officer & Chairman of the Executive Committee, UCB https://www.ucb.com
• Dr Isao Teshirogi, IFPMA Vice President, President and Chief Executive Officer, Shionogi & Co. Ltd. https://www.shionogi.com/global/en/company/history.html
• Albert Bourla, IFPMA Vice President and Vice Chair of the IFPMA CEO Steering Committee; Chairman and Chief Executive Officer, Pfizer
https://www.ifpma.org/who-we-are/our-governance-and-leadership/
2.
Nachweislich bewusste Entscheidung für Impfprodukte trotz vorliegender bedenklicher Studien
Ein ähnliches Konstrukt, die SPEAC, Kooperation zwischen CEPI und der Brighton Collaboration im Jahr 2019, traf die generelle Entscheidung, dass trotz der bekannten Risiken bei Impfungen gegen Coronaviren (ADE) geimpft werden solle, auch mit mRNA, obgleich protokolliert ist, dass man auch von deren besonderen Wirkungen keine Ahnung hat. Das "Forum" hat im Sommer 2020 81 Studien angesehen, die alle gegen die Impfung gegen Coronaviren sprachen, und entschieden, dass man es trotzdem mal versuchen soll. Davor haben sie überprüft, ob Impffolgen der Art, wie sie durch die Studien zu befürchten sind, nachgewiesen werden könnten. Das beruhigende Ergebnis: nein, man kann sie nicht eindeutig nachweisen.
Quellen:
https://brightoncollaboration.us/wp-content/uploads/2021/08/SO2-D2.4_Preliminary-guidance-on-safety-data-collection-for-COVID-19-vaccine-safety_V1.0.pdf
https://brightoncollaboration.us/wp-content/uploads/2021/07/VAED-vaccine-publication.pdf
Zur Geschichte und Bedeutung der Brighton-Collaboration:
https://brightoncollaboration.us/history/
Ihre Entscheidungen wurden von den selben Regulierungsbehörden für verbindlich erklärt, die auch die gleichen Prüfunterlagen der ICH verwenden!
Die Brighton Collaboration Foundation geht auf eine Initiative von Ulrich Heininger zurück. Heininger war und ist Mitglied der RKI-Stiko.
https://brightoncollaboration.us/team/
https://www.rki.de/DE/Content/Kommissionen/STIKO/Mitgliedschaft/Mitglieder/Profile/Heininger_Profil.html
Zum anderen "Hobby" - den PCR-Teststäbchen, hier: deren Qualitätsprüfung, habe ich auch noch ein Schmankerl:
Im Dezember 2021 berichtet die RAPS, die Regulatory Affairs Professionals Society hocherfreut:
Auf erheblichen Druck der Industrie und angesichts der Befürchtungen, dass der Markt für Diagnostika angesichts der COVID-19-Pandemie zusammenbrechen könnte, kündigte die Europäische Kommission im Oktober an, dass sie einige Bestimmungen der IVDR verschieben werde. Zuvor hatte die Kommission als Reaktion auf COVID-19 die Anwendung der Verordnung über Medizinprodukte (MDR) um ein Jahr aufgeschoben. (VERWEIS: IVDR: Kommission schlägt vor, die Umsetzung angesichts des "gravierenden" Mangels an benannten Stellen zu verschieben, Regulatory Focus 14. Oktober 2021)
"Inmitten einer beispiellosen Krise der öffentlichen Gesundheit können wir keine Engpässe bei wichtigen Medizinprodukten riskieren. Die Gesundheitssysteme und die routinemäßigen Gesundheitsdienste sind auf eine nie dagewesene Probe gestellt worden. Die Pandemie hat gleichzeitig den Bedarf an präzisen Diagnosen und einem belastbaren Rechtsrahmen für In-vitro-Medizinprodukte deutlich gemacht. Die Änderung der Verordnung über In-vitro-Diagnostika wird sicherstellen, dass wichtige Medizinprodukte wie COVID oder HIV-Tests weiterhin verfügbar und sicher sind", sagte EU-Gesundheitskommissarin Stella Kyriakides.
Die Änderung sieht vor, die zweijährige Übergangsphase der Verordnung um ein Jahr auf drei Jahre zu verlängern, wobei sich der Übergangszeitraum für die Produkte mit dem geringsten Risiko bis Mai 2027 erstrecken soll, so dass die Testhersteller mehr Zeit haben, ihre Produkte zertifizieren zu lassen, und mehr benannte Stellen ihre Arbeit aufnehmen können, bevor die Verordnung in Kraft tritt.
Die Übergangsfrist für Produkte der Klasse D mit dem höchsten Risiko wird bis Mai 2025 verlängert, während Produkte der Klasse C mit mittlerem Risiko bis Mai 2026 Zeit haben, sich erstmals einer Konformitätsbewertung zu unterziehen. Sterile Diagnostika der Klassen B und A müssen sich bis Mai 2027 einer Konformitätsbewertung unterziehen.
Die Änderung verzögert jedoch nicht das Datum des Inkrafttretens der Verordnung selbst, die nach wie vor am 26. Mai 2022 in Kraft treten soll und in vollem Umfang für CE-gekennzeichnete IVD gilt, die keine Einschaltung einer benannten Stelle erfordern. Andere Aspekte der IVDR, die sich auf die Vigilanz und die Überwachung nach dem Inverkehrbringen beziehen, gelten für alle Produkte, die unter die Verordnung fallen, unabhängig davon, ob sie unter die verlängerte Übergangsfrist fallen.
"Die Entscheidung, die Übergangsbestimmungen der IVD-Verordnung zu ändern, kommt im letzten Moment, um sicherzustellen, dass medizinische Tests für die Patienten und die Gesundheitssysteme in Europa verfügbar bleiben. Es ist nun wichtig, dass die Regulierungsbehörden die verbleibenden kritischen Fragen klären, um die Zertifizierung von mehr als 32.000 medizinischen Tests, die heute auf dem Markt sind, vor Ablauf der neuen Fristen zu ermöglichen, sowie die Zertifizierung innovativer Diagnostika (??) , die sich in der Entwicklung befinden und einen ungedeckten medizinischen Bedarf decken werden", sagte Serge Bernasconi, CEO von MedTech Europe.
MedTech Europe nutzte auch die Gelegenheit, um die EU-Regulierungsbehörden aufzufordern, sich dringend und kontinuierlich mit zwei noch offenen Fragen zu befassen: dem Bedarf an rechtzeitigen Leitlinien zu verschiedenen Aspekten der neuen Verordnung und dem Bedarf an zusätzlichen nach der IVDR benannten Stellen.
Übersetzt mit http://www.DeepL.com/Translator (kostenlose Version)(em>
https://www.raps.org/news-and-articles/news-articles/2021/12/ivdrs-progressive-rollout-gets-official-with-eu-co
Gesagt, getan:
Pressemitteilung20. Dezember 2021Brüssel
Die Änderungsverordnung ändert keine Anforderungen der ursprünglichen In-vitro-Diagnostika-Verordnung (IVD) von 2017. Sie ändert lediglich die Termine für die Anwendung einiger dieser Anforderungen für bestimmte Medizinprodukte.
Für Produkte mit höherem Risiko, wie z. B. HIV- oder Hepatitis-Tests (Klasse D), gelten die neuen Anforderungen ab Mai 2025. Für Produkte der niedrigeren Risikoklasse C, wie z. B. bestimmte Grippetests, wird der Anwendungszeitpunkt bis Mai 2026 verlängert, während für Produkte der niedrigeren Risikoklassen (Sterilität der Klassen B und A) die Anwendung im Mai 2027 beginnt.
Außerdem wird die Anwendung bestimmter Anforderungen für Produkte, die in derselben Gesundheitseinrichtung hergestellt und verwendet werden (so genannte "In-House-Produkte"), um zwei Jahre bis Mai 2024 verschoben. Wenn die Gesundheitseinrichtungen jedoch nachweisen, dass es kein gleichwertiges Produkt auf dem Markt gibt, enden die Übergangsfristen im Mai 2028.
https://ec.europa.eu/commission/presscorner/detail/en/ip_21_6965
Darunter fallen auch die Covid-Tests. Wirklich nett von der Kommission...
Danke, @some.1. Sehr wichtiger Beitrag, den man freilich nicht in 5 Minuten verdauen kann.
Tagesaktuell dringender scheint aber die Auflösung dieses Rätsels:
wo man auf diesem Planeten sehr hohe "Impf"-Quoten hat gibt es rekordverdächtige Infektionszahlen. Wo letztere ungewöhnlich niedrig sind finden wir auch ungewöhnlich niedrige "Impf"-Quoten.
Das kann man nur dadurch erklären dass diese "Impf"-Mittel das Immunsystem in irgendeiner Weise schädigen. Die Frage ist nur: wie? Und: wer könnte das erforschen?
@gelegentlich
Bei der Beurteilung von Wichtigkeit spielt stets auch die "Neigung" eine Rolle. Leider ist Biologie nicht meine Kernkompetenz, selbst wenn mich die Neugier auch hier immer weiter trägt 🙂
Die Verläufe entsprechen ziemlich genau dem, wovor Geert van den Bossche seit Anfang 2021 warnt. Ich neige daher dazu, seinen Erklärungen den höchsten Erkenntniswert zuzuschreiben:
https://www.voiceforscienceandsolidarity.org/blog/scientific-blog?960e36cd_page=3
Um herauszufinden, welche Inhaltsstoffe was wie bewirken, müsste man erst mal zuverlässig wissen, was denn überhaupt genau drin ist und wie viel davon? ALC 01315 und ALC 0159 entsprechen nicht den Anforderungen, schreibt sogar die ansonsten nicht sonderlich kompetente EMA. Es liegen also keine Ergebnisse dazu vor …
Der Meister selbst spricht, dass seine Stoffe bevorzugt dendritische Zellen "anfallen", die Teil des Immunsystems SIND.
https://k.at/news/teil-von-covid-19-vakzine-koennte-aus-oesterreich-kommen/401018798
Es gibt zudem diesen relativ frischen Preprint
https://www.medrxiv.org/content/10.1101/2021.05.03.21256520v1
mit dem sich Jessica Rose
https://i‑do-not-consent.netlify.app/media/Curriculum%20Vitae%20June%202021.pdf
sehr kompetent befasst hat
https://jessicar.substack.com/p/the-bnt162b2-mrna-vaccine-against
Evtl. bringt Sie etwas davon mit Ihrer Frage weiter …
@aa zu "Mit welchen Tricks die Klassifizierung eines Produkts als gentherapeutisch und damit eine strengere Prüfung durch CAT (Committee for Advanced Therapies der EMA) verhindert wird, zeigt dieses Beispiel: […]"
1. Der CAT prüft nicht "strenger" als der CHMP, sondern er wird gutachterlich eingesetzt, wenn es sich um ein ATMP handelt. Die (Nicht-)Empfehlung zur Zulassung an die Europäische Kommission spricht so oder so der CHMP aus, unabhängig davon, ob es sich um ein ATMP handelt oder nicht.
2. In dem konkreten Beispiel ist es ohnehin egal: Denn auch für ein somatisches Zelltherapieprodukt wird der CAT hinzugezogen.
https://www.ema.europa.eu/en/glossary/cat
@Juliane
Die Autoren der Studie (!) haben festgestellt, dass durch die Festlegung der EU RL 2009/120, nach der Impfstoffe gegen Infektionskrankheiten keine Gentherapeutika seien, die Verantwortlichkeit für die PRÜFUNG von der CAT an die CHMP übergeht. Die Kompetenz und die Regularien der beiden sind verscheiden, zu den Aufgaben der CAT gehören auch Gentherapeutika, zu denen der CHMP nicht. Das hat absolut nichts damit zu tun, wer am Schluss mit dem Kopf nickt.
Für die Laien unter uns:
Hier gibt es eine gute Zusammenfassung zu der Frage Gentherapie und/oder Impfung:
https://multipolar-magazin.de/artikel/faktencheck-impfungen-oder-gentherapie
Nina-sangiovese
Eigentlich beschäftigt sich Schreyer mit dieser Frage nur allgemeinsprachlich und setzt sie in einen moralischen Kontext. Das ist ok, ersetzt aber nicht eine Investigation des Regulierungsumfelds. Daher ersetzt das eine nicht das andere.
Mao Kretsch und Hesse sind jetzt "Luca Verweigerer"
https://www.egovernment-computing.de/baden-wuerttemberg-loggt-sich-bei-luca-app-aus-a-1090710/
mRNA-IMPFUNG oder nicht?
Man kann es gar nicht oft genug wiederholen.
Körpereigene Zellen werden veranlasst etwas nicht-körpereigenes zu produzieren. Das basiert auf einer bewusst ausgelösten Manipulation innerhalb körperigener Zellen.
Bei "RNA" handelt es sich um die materielle Basis der Gene. Darum reden wir bei der "mRNA-Impfung" von einer Manipulation. Na und – Legen Hasen Eier? Lasst es sie doch eine Impfung nennen. Bloss, es ist einfach keine.
https://de.wikipedia.org/wiki/Ribonukleins%C3%A4ure
Die aktive Impfung erzeugt eine abgeschwächte Infektion, etwa mit ähnelnden, bewusst injizierten Varianten eines Virus oder eines Bakteriums. Die klassische Impfung.
Die passive Impfung erfolgt durch Gabe von Antikörpern aus Tier oder Mensch.
Eine "indirekte Imunisierungsimpfung" wie durch mRNA ist z.B. in Wikipedia unter Impfung noch gar nicht berücksichtigt worden.
Eine "mRNA-Impfung" ist keine direkte Therapie, weil sie nicht auf einer "Diagnose" beruht. Die präventive T. bei Covid-Erkrankungen erscheint mir jedenfalls albern, sofern die "Zielgruppe" aus relativ gesunden Leuten besteht. "Covid-Erkrankungen" sind im allgemeinen so genannte "Erkältungskrankheiten". Und das seit Jahrtausenden.
Es ist also in der Tat eine Definitionsfrage ob man die mRNA-Impfung eine Impfung nennt oder nicht.
Auf jeden Fall aber handelt es sich um eine nicht notwendige Gen-Therapie. Streng betrachtet also um eine nicht zulässige Behandlung. Logisch, ne?
"Covid", Krankheiten durch Corona-Viren stehen wir wie erwähnt bereits seit Jahrtausenden ohne Impfungen durch. Allermeistens sogar ohne nennenswerte Therapien. Kein Witz! Den besten Schutz bieten warme, dem Wetter entsprechende Kleidung, beheitzte Räume ohne Zugluft und warme Getränke. Die beste Behandlung ist die frühzeitigste. Nicht die modernste.
29.10.2020 / Allgemeine Zeitung (Mainz)
Live-Interview mit Biontech-Chef Prof. Ugur Sahin zu Corona-Impfstoff
Wann ist mit dem Impfstoff zu rechnen? Wie wirksam kann er voraussichtlich sein? Können wir dann endlich zum normalen Leben, wie wir es vor Corona kannten, zurückkehren? Prof. Ugur Sahin, der Vorstandsvorsitzende von Biontech, stellte sich an diesem Donnerstag um 16 Uhr im Live-Interview mit der VRM den Fragen von Chefredakteur Friedrich Roeingh und Reporterchef Christian Matz. Hier sehen Sie die komplette Aufzeichnung.
[ Friedrich Roeingh ]
de.wikipedia.org/wiki/Friedrich_Roeingh
3EuU9lZlycE · ab min 21:42 · eigene Mitschrift
Reporterchef Christian Matz (Allgemeine Zeitung) möchte zu Tozinameran bzw. Comirnaty, damals noch BNT162b2 genannt, wissen:
"Wenn er dann mal da ist, der Impfstoff, wie oft muss man dann zur Impfung, jedes halbe Jahr, einmal im Jahr, alle zwei Jahre?"
"Ja also auch da, das wissen wir noch nicht. Also, ich gehe mal davon aus, dass das nicht jedes halbe Jahr ist. Also, wir können uns da auch an der natürlichen Immunität nach Infektion orientieren. Erst gestern kam in einer Fachzeitschrift, Science, eine Publikation raus, die den Antikörperverlauf nach Infektion sich angeguckt hat. Und was positiv ist, ist, dass dieser Antikörperverlauf nach Infektion eben doch nicht so schnell abnimmt wie das initial spekuliert worden war. Da ist doch bei sehr vielen Probanden auch nach vier bis sechs Monaten nach der Infektion sehr guter Antikörpertiter vorhanden. Der Impfstoff ahmt die natürliche Infektion nach, sodass wir davon ausgehen, dass bei dem Impfstroff auch eine Immunität bestehen wird, die Immunität, die aus meiner Sicht mindestens ein Jahr anhalten wird, und wenn das der kürzeste Zeitraum ist, wo eine Nachimmunisierung notwendig ist, wäre das kein Drama, dann wäre das eine jährliche Nachimmunisierung. Ich gehe davon aus, dass wir wahrscheinlich sogar länger warten können."
— BioNTech-Chef Uğur Şahin am 29. Oktober 2020
https://www.youtube.com/watch?v=3EuU9lZlycE
·
BioNTech-Gründer Uğur Şahin im Interview zu seinem Corona-Impfstoff
02.12.2020 · WELT Nachrichtensender
Großbritannien hat als bisher erstes Land der Welt den Corona-Impfstoff von BioNTech und Pfizer zugelassen. Die zuständige britische Aufsichtsbehörde erteilte eine Notfallzulassung. Der Impfstoff werde ab kommender Woche bereitgestellt, so die britische Regierung. Die EU und die USA müssen dagegen noch auf grünes Licht warten. Im Gespräch dazu ist BioNTech-Gründer Uğur Şahin.
Xx9v3qcC–Y · ab min 8:08 · eigene Mitschrift
WeLT-Moderatorin Marie Przibylla fragt den BioNTech-Chef:
"Können Sie schon abschätzen, wie lange, ich sag mal, die Immunwirkung auch dieses Impfstoffes sein wird? Die Studiendauer war ja jetzt nicht so lang bisher."
"Ja, das ist eine wichtigee Fragestellung. Wir haben momentan Daten von circa drei Monaten nach Immunisierung. Wir werden weiter Daten, und zwar für über zwei Jahre sammeln. Was wir momentan sehen ist, dass die Immunantwort auch nach drei Monaten sehr stark ausgeprägt ist. Und was wir ja auch an Patienten, die COVID-19 durchgemacht haben, beobachten können ist, dass Patienten, die eine hohe Immunantwort gebildet haben, hohe Antikörper-Immunantwort ausgebildet haben, auch nach sechs Monaten diese Immunantwort haben. So dass wir davon ausgehen, dass unser Impfstoff, der ja eine noch stärkere Immunantwort ausbildet als die Krankheit selber, sehr wahrscheinlich uns helfen wird, zumindest mal ein Jahr eine Kontrolle bereit zu stellen. Aber wie gesagt, diese Daten müssen wir generieren und diese Daten werden vorliegen. Und wenn wir dann feststellen sollten, dass man noch mal nachimmunisieren muss, ist das auch kein Problem, das ist ja nicht unüblich, dass Impfstoffe in Intervallen dann auch wiederholt gegeben werden."
— Uğur Şahin am 2. Dezember 2020
https://www.youtube.com/watch?v=Xx9v3qcC–Y
·
There is no pandemic, there is COVAX, a crime against humanity and a medical crime. “STOP COVAX”